Данное сообщение я хотел бы посвятить обзору формата файлов GFF, который очень широко используется в биоинформатике для структурирования, хранения и передачи данных о функциональных элементах генома. Это сообщение будет начинать серию рассказов не только о спецификации структуры данных вида GFF, но и о загрузке файлов формата GFF в рабочее пространство R среды, преобразовании их в объекты классов GRanges и TxDb, анализу структуры таких объектов и их использованию в исследованиях транскриптома.
Формат файлов GFF (акроним от англ. General Feature Format, либо реже используемых в настоящее время Gene-Finding Format или Generic Feature Format) – один из способов организации, хранения, обмена и представления данных о признаках (характеристиках или функциональных элементах) генома. При этом под признаками генома подразумеваются сигнальные последовательности (стартовые кодоны, стоп-кодоны, мотивы, сайты сплайсинга и т. д.), а также протяженные сегменты (промоторные регионы, экзоны, интроны и т. д.), которые могут быть скомбинированы в целые гены, РНК или белковые продукты.
Первоначальная версия формата GFF была предложена Dr. Richard Durbin из Института Сенгера (Кембриджшир, Великобритания) и Dr. David Haussler из Калифорнийского университета (Санта Круз, США). Однако из-за ряда недостатков первоначальная версия вскоре была модифицирована и расширена до версий GFF2 и GFF3, которые являются современными стандартами формата GFF. Вне зависимости от версии файл в формате GFF является текстовым файлом, где для каждого признака генома отводится одна строка, каждая строка содержит 9 полей, разделенных знаком табуляции, а всей области данных предшествует область комментариев. Такая структура файла позволяет быстро извлекать необходимые данные и проводить их обработку с помощью соответствующих программных средств.
Далее мы рассмотрим структуру файлов формата GFF2, GFF2.5 и GFF3; формат же GFF1 мы оставим без внимания, так как он во многом уже потерял актуальность.
Описание формата GFF2.
Область комментариев.
Строки с комментариями начинаются с одного или двух символов #. Строки, начинающиеся с одного символа #, содержат информацию только для пользователя и игнорируются программой, проводящей парсинг (разбор или анализ) файла GFF2. Cтроки, начинающиеся с двух символов #, содержат информацию о всем файле и именуются директивами (псевдокомментариями или метаданными). Они не являются обязательными, но, как правило, присутствуют в любом файле GFF2, так как используются программой, осуществляющей парсинг данных. В этих строках содержится следующая информация.
##gff–version 2
Версия формата GFF. Эта строка располагается первой и в файле встречается только один раз.
##source-version <source> <version text>
Наименование и версия программы, которая была использована при создании файла GFF2.
##date <date>
Дата создания файла GFF2.
##Type <type> [<seqname>]
Тип последовательности (“DNA”, “RNA” или “Protein”), признаки которой содержатся в файле. В одном и том же файле могут быть представлены разные типы последовательностей. Если этот комментарий отсутствует, то по умолчанию принимается, что тип последовательности – ДНК.
##DNA <seqname>
Последовательность ДНК вида:
##acggctcggattggcgctggatgatagatcagacgac
##…
##end–DNA
Используется редко.
##RNA <seqname>
Последовательность РНК вида:
##acggcucggauuggcgcuggaugauagaucagacgac
##…
##end–RNA
Используется редко.
##Protein <seqname>
Последовательность белка вида:
##MVLSPADKTNVKAAWGKVGAHAGEYGAEALERMFLSF
##…
##end–Protein
Используется редко.
##sequence-region <seqname> <start> <end>
Комментарий, показывающий, что в файле содержатся признаки только указанного региона.
Область данных.
Как уже говорилось, эта область представлена множеством строк (по одной на каждый признак), а каждая строка содержит 9 полей, разделенных знаком табуляции. Список полей и их содержание приведены ниже.
Поле 1: <seqname>
Название последовательности. Обычно имя последовательности соответствует идентификатору этой последовательности в сопроводительном файле формата FASTA, либо идентификатору этой последовательности в базе данных (например, это может быть номер доступа EMBL/GenBank/DDBJ).
Поле 2: <source>
Источник текущего признака (программа, база данных, экспериментальные данные и т. д.).
Поле 3: <feature>
Тип признака (например, “gene”, “transcript”, “CDS”, “ncRNA”).
Поле 4: <start>
Целочисленное значение координаты начала признака. При этом нумерация всей последовательности начинается с 1, а начальная координата признака должна быть меньше или равна конечной координате.
Поле 5: <end>
Целочисленное значение координаты конца признака.
Поле 6: <score>
Дробное число с плавающей точкой, показывающее, насколько надежно идентифицирован признак.
Поле 7: <strand>
Цепь, которой принадлежит признак. Может принимать три значения: “+”, “–“ или “.”. Последний вариант используется тогда, когда информация о цепи не является значимой (например, в случае динуклеотидных повторов).
Поле 8: <frame>
Поле содержит информацию об отношении признака к открытой рамке считывания. Это поле может принимать одно из четырех значений: “0”, “1”, “2” или “.”. Значение “0” указывает на то, что признак находится внутри открытой рамки считывания и первый нуклеотид этого признака соответствует первому нуклеотиду кодона. Значение “1” указывает на наличие одного, а значение “2” на наличие двух “лишних” нуклеотидов у признака, в результате чего только его второй или третий нуклеотиды, соответственно, начинают кодон. Значение “.” используется для заполнения поля <frame> признаков РНК и белков.
Поле 9: [attribute]
Поле содержит множество различных атрибутов признака, собранных в одну группу. Такими атрибутами могут быть идентификаторы, классы биологических молекул, бэнды хромосом, альтернативные названия признака, названия синдромов и т. д. Атрибуты собраны в одну строку и разделены точкой с запятой. Один и тот же атрибут может иметь несколько значений, разделенных запятой.
Формат GFF2 имеет ряд недостатков, в частности, он может представлять только двухуровневые иерархии признаков, и, соответственно, не может справиться с трёхуровневой иерархией типа ген → транскрипт → экзон. Более того, даже в случае с двухуровневой иерархией в формате GFF2 не предусмотрена запись направления этой иерархии. Эти, а также некоторые другие недостатки формата GFF2 привели к появлению его уточненной версии GFF2.5, а также более совершенного формата GFF3. Более подробно о формате GFF2 можно узнать на официальном сайте Института Сенгера, сотрудники которого являются разработчиками данного стандарта организации, хранения, обмена и представления данных о функциональных элементах генома.
Описание формата GFF2.5.
Формат GFF2.5 чаще всего называется форматом GTF (акроним от англ. Gene Transfer Format), так как он является расширением формата GFF2. В этом формате первые 8 полей идентичны таковым в формате GFF2, а вот поле “attribute” включает два обязательных атрибута, предназначенных для обработки нескольких транскриптов и одной и той же области генома.
gene_id value
Уникальный идентификатор геномного локуса, откуда происходит транскрипт.
transcript_id value
Уникальный идентификатор транскрипта.
Другие атрибуты могут быть произвольными.
Описание формата GFF3.
Формат GFF3 учитывает те недостатки, которые есть в предыдущих версиях формата GFF. В частности, в этом формате предусмотрена многоуровневая иерархия признаков, допускается одновременная принадлежность одного и того же признака разным группам, решена проблема записи признаков, занимающих разные регионы и т. д.
Область комментариев.
Строки с комментариями начинаются с одного, двух или трех символов #. Строки, начинающиеся с одного символа #, содержат информацию только для пользователя и игнорируются программой, проводящей парсинг файла GFF3. Программа так же игнорирует пустые строки или строки, которые заканчиваются на символ #. Cроки, начинающиеся с двух символов #, являются директивами и, как правило, используются программой парсинга. В этих строках может быть записана информация следующего рода.
##gff–version *
Версия формата GFF. Эта строка располагается первой, всегда присутствует и в файле встречается только один раз. В случае с файлами формата GFF3 символ * всегда имеет значение 3, как показано но нижеследующем примере.
##gff-version 3
##sequence-region seqid start end
Информация об опорной последовательности, присутствующей в области данных. Записывается в формате “seqid start end”, не является обязательной, но желательной, так как может использоваться программой парсинга при проверке границ признаков. Поскольку опорных последовательностей в области данных может быть много, то и строк ##sequence–region seqid start end так же может быть много (по одной на каждую опорную последовательность, без повторов и без перекрестных ссылок). При этом следует иметь ввиду, что наличие строк ##sequence–region seqid start end для всех последовательностей в области данных не обязательно, но если строка имеется, то в области данных обязательно должна присутствовать соответствующая последовательность (исключением могут быть только признаки, помеченные атрибутом “Is_circular”). Ниже дано несколько примеров записи такого рода информационных строк:
##sequence-region 10 1 133797422
##sequence-region KI270757.1 1 71251
##sequence-region MT 1 16569
##sequence-region X 1 156040895
##sequence-region Y 2781480 56887902
##feature–ontology URI
Информация о том, что в файле GFF3 тип признака определен по данным онтологического анализа, которые доступны по указанным URI или URL. Комментарий добавлен в спецификации файлов GFF3 недавно и пока широко не используется.
##attribute–ontology URI
Информация о том, что в файле GFF3 имена атрибутов взяты из данных онтологического анализа, которые доступны по указанным URI или URL. Комментарий добавлен в спецификации файлов GFF3 недавно и пока широко не используется.
##source–ontology URI
Информация о том, что в файле GFF3 имена источников взяты из данных онтологического анализа, которые доступны по указанным URI или URL. Комментарий добавлен в спецификации файлов GFF3 недавно и пока широко не используется.
##species NCBI_Taxonomy_URI
Информация о том, к какому виду живых организмов относятся данные в файле GFF3. Как правило, это URL-адрес страницы, посвященной данному виду, в таксономическом обозревателе NCBI, записанный в одном из двух вариантов:
##species http://www.ncbi.nlm.nih.gov/Taxonomy/Browser/wwwtax.cgi?id=6239
##species http://www.ncbi.nlm.nih.gov/Taxonomy/Browser/wwwtax.cgi?name=Caenorhabditis+elegans.
##genome–build source buildName
Источник и имя сборки генома, по которой в файле GFF3 даны координаты всех признаков. Ниже дано несколько примеров:
##genome–build NCBI B36
##genome–build WormBase ws110
##genome–build FlyBase r4.1
Следует отметить, что начиная с версии 1.20 спецификаций GFF3 информация о геноме может быть разнесена по нескольким строкам с указанием имени сборки, версии, даты, источника и кода доступа:
#!genome-build GRCh38.p3
#!genome-version GRCh38
#!genome-date 2013-12
#!genome-build-accession NCBI:GCA_000001405.18
В таких случаях в начале строки с комментариями используется не двойной символ ##, а два символа #!, так как это пока экспериментальное нововведение.
##FASTA
Информация о том, что файл содержит нуклеотидные или белковые последовательности в формате FASTA. Если имеются последовательности, то все они собираются вместе и помещаются в конец файла, как на примере, представленном ниже:
##FASTA
>ctg123
CTTCTGGGCGTACCCGATTCTCGGAGAACTTGCCGCACCATTCCGCCTTG
TGTTCATTGCTGCCTGCATGTTCATTGTCTACCTCGGCTACGTGTGGCTA
TCTTTCCTCGGTGCCCTCGTGCACGGAGTCGAGAAACCAAAGAACAAAAA
AAGAAATTAAAATATTTATTTTGCTGTGGTTTTTGATGTGTGTTTTTTAT
AATGATTTTTGATGTGACCAATTGTACTTTTCCTTTAAATGAAATGTAAT
…
Строки, начинающиеся с трех символов #, сигнализируют о том, что все данные, которые располагались выше этой строки, полностью обработаны. В этом случае программа, осуществляющая парсинг, может закрыть все вспомогательные объекты, которые до сих пор использовались в обработке файла и приступить к работе со следующим диапазоном данных.
Кроме описанных комментариев допускается включение и другой информации. Так, например, область комментариев нередко содержит информацию об использованных при создании файла GFF3 программных средствах, сроках и источниках аннотирования (процессе маркировки генов и других признаков генома), а также другие сведения:
#!processor NCBI annotwriter
#!annotation-date 12 March 2015
#!annotation-source NCBI Homo sapiens Annotation Release 107
Область данных.
Как и в случае с другими версиями формата GFF, эта область представлена множеством строк (по одной на каждый признак), а каждая строка содержит 9 полей, разделенных знаком табуляции.
Поле 1: “seqid“
Метка системы координат (например, хромосомы), по которой можно определить местоположение текущего признака.
Поле 2: “source“
Наименование алгоритма или процедуры, с помощью которой был идентифицирован признак. Обычно это название программы или ее части (например, “havana“, “Gnomon”), либо наименование базы данных (например, “GenBank”).
Поле 3: “type“
Тип признака (например, “gene”, “transcript”, “CDS”, “ncRNA”).
Поле 4: “start“
Начальная координата признака в геноме (в системе координат, помеченной в поле 1). Координата всегда имеет положительное целочисленное значение, меньшее либо равное значению координаты конца текущего элемента. Начальная координата признаков с так называемой нулевой длиной (например, инсерции) всегда равна конечной координате, а сам признак будет располагаться справа от этой координаты по направлению системы координат.
Поле 5: “end“
Конечная координата признака в геноме (в системе координат, помеченной в поле 1). Координата всегда имеет положительное целочисленное значение, большее либо равное значению координаты начала текущего признака.
Поле 6: “score“
Дробное число с плавающей точкой, показывающее, насколько надежно идентифицирован признак. К сожалению, однозначного подхода в расчете этого параметра нет и чаще всего в файлах GFF3 значения этого поля заменены на символ периода “.”.
Поле 7: “strand“
Цепь, в которой локализован признак: “+” – плюс цепь, “–” – минус цепь, “?” – информация о цепи значима, но неизвестна, “.” – информация о цепи незначима.
Поле 8: “phase“
Поле заполняется только для признаков “CDS” и может иметь только одно из трех целочисленных значений: 0, 1 или 2. Эти значения указываю на то, сколько нужно убрать нуклеотидов с самого начала признака, что бы достигнуть первого нуклеотида следующего кодона. Добавим, что для признаков, локализованных в плюс цепи, фаза считается от начала признака, а для признаков из минус цепи – с конца признака.
Поле 9: “attributes“
Список атрибутов признака в формате “бирка = значение” (“tag = value”). Если у одного признака много атрибутов, то они отделяются друг от друга точкой с запятой; если же имеется много значений одного и того же атрибута, то эти значения разделяют запятой. Ниже перечислены и описаны теги с предопределенными значениями, но в некоторых файлах могут встречаться и другие атрибуты.
ID
Уникальный идентификатор признака. Один и тот же идентификатор может многократно повторятся в одном файле, но только в том случае, когда он представляет прерывистый признак (один признак, существующий во множестве генетических локусов). В таких случаях признак представлен не одной строкой, а совокупностью строк.
Name
Имя признака. Не всегда уникально даже в случае с непрерывными признаками.
Alias
Альтернативное имя признака. Не всегда уникально даже в случае с непрерывными признаками.
Parent
Тег, обозначающий “родителя” признака. Такого рода тег используется для группировки признаков низкого уровня в признаки более высокого уровня, например, экзонов в транскрипты, транскриптов в гены и т. д. Отметим, что признак может иметь много “родителей”.
Target
Идентификатор и локализация нуклеотидной или аминокислотной последовательности, относительно которой проводилось выравнивание последовательности признака. Этот тег записывается в формате “target_id start end [strand]”, причем цепь указывается не всегда.
Gap
Пометка о том, что последовательность признака и целевая последовательность, относительно которой проводилось выравнивание, не коллинеарны (содержат пробелы).
Derives_from
Тег используется для обозначения признаков, принадлежащих полицистронным генам.
Note
Пометка произвольного содержания. Пометок у одного признака может быть много.
Dbxref
Перекрестная ссылка на базу данных. Может иметь множество значений.
Ontology_term
Перекрестная ссылка на онтологический термин. Может иметь множество значений.
Is_circular
Тег, обозначающий, является ли признак кольцевым.
Ниже представлен пример организации данных о кодирующем белок гене EDEN человека в формате GFF3. В верхней части рисунка мы видим структуру гена EDEN, отображаемую в геномном браузере UCSC Genome Browser. А далее идет информация об этом же гене, но уже в формате GFF3.

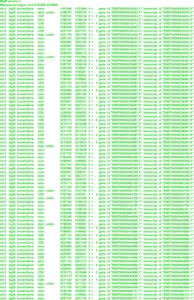
Более подробно о формате GFF3 можно узнать на официальном сайте международного проекта The Sequence Ontology Project, исполнители которого являются разработчиками данного стандарта организации, хранения, обмена и представления данных о функциональных элементах генома.
Практическое применение формата GFF.
Формат GFF может быть использован для решения очень разнообразных задач биоинформатики, молекулярной и системной биологии. Ниже перечислены лишь некоторые из таких задач:
- обмен стандартизированными данными внутри научного сообщества;
- представление экспериментальных данных в стандартизированном виде, готовом к дальнейшему анализу;
- тестирование методов идентификации функциональных элементов генома;
- поиск новых, более сложных функциональных элементов генома с использованием стандартизованных данных, полученных их разных источников или же содержащих разные характеристики признаков;
- подача стандартизированных данных в программы, осуществляющих пост-анализ;
- подача стандартизированных данных в программы визуализации предсказанных и экспериментально подтвержденных признаков генома.