Наша команда предлагает различные варианты информационно-консультативного сотрудничества в области анализа биомедицинских данных. Оказываем помощь в анализе больших биологических и медицинских данных, их визуализации и интерпретации. Работаем с данными любой сложности (в том числе с неструктурированными данными, пропусками и зашумлением), арсенал аналитических инструментов – от стандартных статистических методов до алгоритмов машинного обучения, визуализация – во всем ее разнообразии.
Мы выполняем следующие виды анализа:
- Стандартный статистический анализ данных
- Геномика
- Транскриптомика
- Идентификация и анализ малых кэппированных РНК
- Оценка скорости работы РНК полимеразы II млекопитающих
- Идентификация и аннотация экспрессирующихся генов
- Анализ дифференциальной экспрессии генов
- Идентификация сайтов РНК-связывающих белков в разных классах РНК
- Сборка транскриптома de novo
- Сборка транскриптома с опорой на эталонные аннотации
- Идентификация и аннотация сохраненных интронов
- Анализ дифференциального сплайсинга
- Идентификация и аннотация открытых рамок считывания в полноразмерных молекулах РНК
- Реконструкция, топологический и функциональный анализ сетей генных регуляций
- Одномерный анализ выживаемости больных по транскриптомным данным
- Многомерный анализ выживаемости больных по транскриптомным данным
- Построение классификационных и предиктивных моделей на основе мета-классификаторов и транскриптомных данных
- Эпигеномика
- Протеомика
- Графическая визуализация данных
Стандартный статистический анализ данных.
Проведение типового статистического анализа двух (или большего количества) выборок, нацеленного на установление различий между этими выборками. Анализ включает следующие этапы:
- разведочный анализ данных;
- оценка характера распределения данных;
- описательная статистика данных;
- проверка нулевой гипотезы;
- визуализация и интерпретация результатов анализа.
Идентификация полиморфных сайтов по данным направленного экзомного секвенирования.
Анализ нацелен на идентификацию полиморфных сайтов в ограниченной выборке экзонов целевых генов, прочитанных с помощью направленного (таргетированного) экзомного секвенирования. Анализ включает следующие этапы:
- оценка качества первичных данных;
- предпроцессинг и фильтрация первичных данных;
- картирование коротких прочтений;
- идентификация полиморфных сайтов;
- аннотация идентифицированных полиморфных сайтов;
- визуализация и интерпретация результатов анализа.
Идентификация полиморфных сайтов по данным полноэкзомного секвенирования.
Анализ нацелен на идентификацию полиморфных сайтов во всех экзонах полного списка генов, прочитанных с помощью полноэкзомного секвенирования. Анализ включает следующие этапы:
- оценка качества первичных данных;
- предпроцессинг и фильтрация первичных данных;
- картирование коротких прочтений;
- идентификация полиморфных сайтов;
- аннотация идентифицированных полиморфных сайтов;
- визуализация и интерпретация результатов анализа.
Идентификация полиморфных сайтов по данным полногеномного секвенирования.
Анализ нацелен на идентификацию полиморфных сайтов по данным полногеномного секвенирования, включая межгенные области и интронные последовательности. Анализ включает следующие этапы:
- оценка качества первичных данных;
- предпроцессинг и фильтрация первичных данных;
- картирование коротких прочтений;
- идентификация полиморфных сайтов;
- аннотация идентифицированных полиморфных сайтов;
- визуализация и интерпретация результатов анализа.
Оценка скорости работы РНК полимеразы II.
РНК полимераза II является ключевой полимеразой, осуществляющей транскрипцию генов эукариот. Скорость работы этой полимеразы зависит от ряда факторов (pH среды, присутствие белковых и небелковых регуляторов транскрипции, структура хроматина и т. д.), изучение которых имеет как фундаментальное, так и прикладное значение, в частности, при разработке высокоселективных ингибиторов, эпигенетических модуляторов и других молекулярных терапевтиков. Предлагаемый метод оценки скорости работы РНК полимеразы II основан на использовании данных, получаемых с помощью метода 4sU DRB-Seq. Этот метод включает блокаду активности клеточных РНК полимераз II в момент их перехода от инициации транскрипции к элонгации, из-за чего молекулы полимеразы накапливаются в 5’UTR областях генов. После снятия блокировки наблюдается «волна» транскрипции, которая детектируется с помощью полнотранскриптомного секвенирования. Геномные координаты «волн» такой транскрипции определяются с помощью скрытых моделей Маркова. Если данные секвенирования получены по нескольким временным точкам, начиная с момента снятия блокировки, то это позволяет методом линейной регрессии рассчитать скорость транскрипции индивидуальных генов, осуществляемой РНК полимеразой II, что проиллюстрировано на нижеследующем рисунке.

Обзор метода 4sUDRB-Seq для оценки скорости работы РНК полимеразы II. a) Формирование «волны» транскрипции в гене TOP1 человека после деблокирования РНК полимеразы II. б) Определение границ «волны» транскрипции с помощью скрытой модели Маркова с тремя состояниями. с) Расчет скорости работы РНК полимеразы II методом линейной регрессии.
В целом, биоиформатический анализ 4sU DRB-Seq данных включает следующие этапы:
- разработка транскрипционных моделей генов;
- оценка качества первичных 4sU DRB-Seq данных;
- предпроцессинг и фильтрация первичных 4sU DRB-Seq данных;
- картирование коротких прочтений, полученных методом 4sU DRB-Seq;
- суммаризация коротких прочтений и формирование первичной матрицы подсчетов;
- фильтрация первичной матрицы подсчетов;
- нормализация 4sU DRB-Seq библиотек и трансформация данных;
- идентификация геномных координат «волн» активности РНК полимеразы II с помощью скрытых моделей Маркова с тремя состояниями;
- расчет скорости работы РНК полимеразы II с помощью линейной регрессии;
- визуализация и интерпретация результатов анализа.
Боле подробно о методе можно почитать в работе Danko C. G. с соавторами (Danko C. G., Hah N., Luo X., Martins A. L., Core L., Lis J. T., Siepel A., Kraus W. L. Signaling pathways differentially affect RNA polymerase II initiation, pausing, and elongation rate in cells. // Molecular Cell. – 2013. – Vol. 25. – P. 212-222), а также в одной из наших последних публикаций (Radzisheuskaya A., Shliaha P. V., Grinev V. V., Shlyueva D., Koche R., Gorshkov V., Kovalchuk S., Zhan Y., Rodriguez K. L., Johnstone A. L., Keogh M.-C., Hendrickson R. C., Jensen O. N., Helin K. Complex-dependent histone acetyltransferase activity of KAT8 determines its role in transcriptional regulation and cellular homeostasis. // Molecular Cell. – 2021. – Vol. 81. – P. 1749-1765).
Идентификация и аннотация экспрессирующихся генов.
В пайплайне реализован стандартный подход по оценке экспрессии генов, основанный на полнотранскриптомном секвенировании. Анализ включает следующие этапы:
- подготовка эталонной (референсной) последовательности целевого генома;
- расчет хеш-таблицы (индексация эталонного генома);
- оценка качества первичных данных;
- предпроцессинг и фильтрация первичных данных;
- картирование коротких прочтений;
- суммаризация коротких прочтений и формирование первичной матрицы подсчетов;
- фильтрация первичной матрицы подсчетов;
- формирование списка активных генов;
- аннотация экспрессированных генов;
- визуализация и интерпретация результатов анализа.
Анализ дифференциальной экспрессии генов.
Типовой пайплайн, основанный на линейном моделировании, для идентификации генов, дифференциально экспрессированных в двух (или более) условиях. Анализ включает следующие этапы:
- оценка качества первичных данных;
- предпроцессинг и фильтрация первичных данных;
- картирование коротких прочтений;
- суммаризация коротких прочтений и формирование первичной матрицы подсчетов;
- фильтрация первичной матрицы подсчетов;
- нормализация RNA-Seq библиотек;
- трансформация данных и устранение их гетероскедастичности;
- линейное моделирование;
- байесовская статистика и идентификация дифференциально экспрессированных генов;
- аннотация дифференциально экспрессированных генов;
- визуализация и интерпретация результатов анализа.

Типовой результат анализа дифференциальной экспрессии генов, представленный в виде графиков-вулканов. Графики воспроизведены из нашей недавней работы Radzisheuskaya A., Shliaha P. V., Grinev V. V., Shlyueva D., Koche R., Gorshkov V., Kovalchuk S., Zhan Y., Rodriguez K. L., Johnstone A. L., Keogh M.-C., Hendrickson R. C., Jensen O. N., Helin K. Complex-dependent histone acetyltransferase activity of KAT8 determines its role in transcriptional regulation and cellular homeostasis. // Molecular Cell. – 2021. – Vol. 81. – P. 1749-1765
Идентификация и аннотация сохраненных интронов.
Пайплайн позволяет идентифицировать три класса интронов, сохраняющихся в молекулах РНК во время сплайсинга: экзитроны (exitrons), удержанные (retained) и задержанные (detained) интроны. В качестве вводных в пайплайне используются данные полнотранскриптомного секвенирования. Анализ включает следующие этапы:
- оценка качества первичных данных полнотранскриптомного секвенирования;
- предпроцессинг и фильтрация первичных данных;
- картирование коротких прочтений;
- разработка списка геномных координат неперекрывающихся интронных бинов;
- расчет эффективной длины интронных бинов по матрицам отображаемости;
- суммаризация коротких прочтений по интронным бинам и формирование первичной матрицы подсчетов;
- фильтрация первичной матрицы подсчетов;
- преобразование первичной матрицы подсчетов, стабилизирующее дисперсию;
- расчет нулевой модели распределения подсчетов по интронным бинам;
- трансформация данных и устранение их гетероскедастичности;
- линейное моделирование;
- байесовская статистика и идентификация интронов, дифференциально экспрессированных по сравнению с нулевым распределением;
- фильтрация, отбор и аннотация сохраненных интронов;
- визуализация и интерпретация результатов анализа.
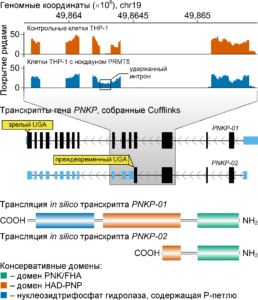
Пример идентификации интронов, сохраняемых при процессинге молекул РНК генов человека. Рисунок воспроизведен из нашей недавней работы Radzisheuskaya A., Shliaha P. V., Grinev V. V., Lorenzini E., Kovalchuk S., Shlyueva D., Gorshkov V., Jensen O. N., Helin K. PRMT5 methylome profiling uncovers a direct link to splicing regulation in human acute myeloid leukemia. // Nature Structural and Molecular Biology. – 2019. – Vol. 26. – № 11. – P. 999-1012. – DOI: 10.1038/s41594-019-0313-z
Одномерный анализ выживаемости больных по транскриптомным данным.
Пайплайн нацелен на изучение влияния отдельных генов (ковариат) на выживаемость людей, страдающих тем или иным заболеванием (сердечно-сосудистая патология, онкологические заболевания, сахарный диабет и т. д.). В пайплайне используются транскриптомные данные или же данные по экспрессии целевых генов, полученные с помощью классических методов молекулярной биологии (например, количественной ОТ-ПЦР), а также правосторонние цензурированные данные по клиническому исходу (общая выживаемость, выживаемость без прогрессии и т. д.). Эффект изучаемого гена на выживаемость оценивается с помощью одномерной регрессионной модели пропорциональных рисков Кокса. Анализ включает следующие этапы:
- подготовка клинических данных;
- подготовка данных по экспресии изучаемых генов;
- фильтрация, нормализация и трансформация первичных данных;
- расчет параметров одномерной регрессионной модели пропорциональных рисков Кокса;
- обобщение статистических характеристик модели;
- графическая визуализация результатов анализа с помощью кривой Каплана-Мейера;
- интерпретация результатов.
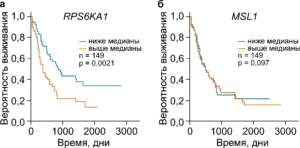
Визуальное представление выживаемости больных острым миелоидным лейкозом в зависимости от экспресии генов RPS6KA1 (а) или MSL1 (б). Кривые Каплана-Мейера воспроизведены из нашей недавней работы Grinev V. V., Barneh F., Ilyushonak I. M., Nakjang S., Smink J., van Oort A., Clough R., Seyani M., McNeill H., Reza M., Martinez-Soria N., Assi S. A., Ramanouskaya T. V., Bonifer C., Heidenreich O. RUNX1/RUNX1T1 mediates alternative splicing and reorganizes the transcriptional landscape in leukemia. // Nature Communications. – 2021. – Vol. 12(1). – doi: 10.1038/s41467-020-20848-z
Построение классификационных и предиктивных моделей на основе мета-классификаторов и транскриптомных данных.
В основе предлагаемого пайплайна лежит использование мета-классификаторов типа метода опорных векторов, “случайного леса” из деревьев принятия решения, градиентного бустинга и т. д. (выбор метода(-ов) определяется решаемой задачей) для построения моделей нелинейных взаимосвязей между переменной-откликом (например, выживаемостью пациентов) и признаками-предикторами (например, экспрессией генов в целевых клетках пациента). Такого рода модели позволяют идентифицировать предикторы (гены), значимо связанные с переменной-откликом (провести так называемый отбор признаков, или features selection), и выстраивать прогноз исхода (например, выживаемости пациентов или их отклика на тот или иной протокол лечения). Анализ включает следующие этапы:
- предварительная подготовка транскриптомных данных (внедрение фиктивных переменных, удаление предикторов с нулевой или близкой к нулевой дисперсией, удаление сильно коррелируемых предикторов, центрирование и шкалирование данных);
- стратифицированное (районированное) разбиение выборки на обучающую и проверочную под-выборки;
- расчет гиперпараметров классификационной модели и ее кросс-валидация;
- оценка качества классификационной модели на проверочной под-выборке;
- оценка значимости предикторов, их ранжирование и отбор;
- построение прогноза на новых данных, визуализация и интерпретация результатов.
Примеры успешного использования такого рода подходов для решения фундаментальных задач можно найти в наших недавних работах (Grinev V. V., Barneh F., Ilyushonak I. M., Nakjang S., Smink J., van Oort A., Clough R., Seyani M., McNeill H., Reza M., Martinez-Soria N., Assi S. A., Ramanouskaya T. V., Bonifer C., Heidenreich O. RUNX1/RUNX1T1 mediates alternative splicing and reorganizes the transcriptional landscape in leukemia. // Nature Communications. – 2021. – Vol. 12(1). – doi: 10.1038/s41467-020-20848-z; Grinev V. V., Nazarov P. V., Klimov E. A. Power-law behavior of alternative splicing of exons in human transcriptome. // Annual Research & Review in Biology. – 2019. – Vol. 32, Issue 6. – P. 1-13. DOI: 10.9734/arrb/2019/v32i630105; Grinev V. V., Migas A. A., Kirsanava A. D., Mishkova O. A., Siomava N., Ramanouskaya T. V., Vaitsiankova A. V., Ilyushonak I. M., Nazarov P. V., Vallar L., Aleinikova O. V. Decoding of exon splicing patterns in the human RUNX1-RUNX1T1 fusion gene. // The International Journal of Biochemistry and Cell Biology. – 2015. – Vol. 68. – P. 48-58).
Графическая визуализация данных.
Построение графиков высокого качества является неотъемлемой и очень важной частью работы с первичными данными и результатами их анализа. Особенно это актуально при работе с большими многомерными биологическими и медицинскими данными, а также при подготовке результатов исследования к публикации в высокорейтинговых научных журналах.
Мы предлагаем помощь в создании графиков любой сложности. Основной (но не единственный) инструмент – графические возможности статистической среды программирования R, включая базовую систему построения графиков, а также системы grid, lattice и ggplot2. Графики могут быть сохранены в любом векторном (*.pdf, *.wmf, *.svg и др.) или растровом (*.jpeg, *.tiff, *.png и др.) форматах. При построении графиков учитываются эстетические характеристики рисунка, а также требования по цветовой гамме с учетом цветоаномалий зрения некоторых людей.
О графических возможностях статистической среды программирования R вы можете узнать из нашей вводной лекции, доступной на образовательном YouTube канале Grinev’s Educational Channel. Коллекция наших рисунков доступна на странице BioData Analytics в Instagram. Кроме того, разнообразные рисунки были использованы нами в наших публикациях и также доступна для изучения.